http://www.webciencia.com/
http://www.bussolaescolar.com.br/
http://www.enaol.com/index.php
http://noticias.terra.com.br/educacao/
Além dos outros links, postados após as publicações.
quinta-feira, 26 de julho de 2012
quarta-feira, 25 de julho de 2012
Conclusões
O avanço científico traz consigo benefícios nunca antes imaginados. Parece ser questão de tempo obtermos uma cura descoberta por meios que nem esperávamos ser possível. Quando poderíamos imaginar que poderíamos modificar geneticamente um alimento, com o fim de beneficiar não somente nossa saúde, mas o cultivo e a comercialização? Ou como poderíamos fantasiar um ser idêntico a outro ser, derivado dele próprio? Muito provavelmente não tão cedo.
Durante este trabalho, me atentei para as peculiaridades. Mesmo antes, nas síndromes e doenças. Parece-me incrível como um cromossomo a menos (ou a mais) pode causar tantas mudanças no desenvolvimento de um ser. E lá vem de novo o avanço científico, de como hoje em dia sabemos os detalhes, as causas e curas, as prevenções, e tudo o mais.
Contei também com um enriquecimento de informações. Muitos dos casos pesquisados neste trabalho eu não sabia ou sabia por alto, sem ter o conhecimento, muitas vezes, das suas principais características. E essas, me referindo às síndromes, na maioria das vezes são estéticas, o que de primeira nos causa um impacto, seguido de um olhar curioso. Pior são aquelas prejudiciais à saúde do indivíduo, o que inconscientemente nos desperta um sentimento de compaixão, consentimento instantâneo.
No fim da pesquisa, pude notar as diversidades dos casos discorridos. A riqueza de detalhes que é a biologia, a biotecnologia, a medicina, a vida, num todo. Às vezes, detalhes complexos demais para um entendimento amplo e total, mas capaz de mesmo assim nos proporcionar um acréscimo no conhecimento, o que com toda a certeza foi o objetivo principal do portfólio.
E como se não bastasse tudo isso, é notório a todos que ainda há muito o que se descobrir. A medicina tem muito a evoluir, os cientistas a se aprimorar, a genética, a ser mais utilizada e aproveitada...
Foi certamente um trabalho produtivo e os assuntos pesquisados, úteis para o nosso entendimento de vida .
Durante este trabalho, me atentei para as peculiaridades. Mesmo antes, nas síndromes e doenças. Parece-me incrível como um cromossomo a menos (ou a mais) pode causar tantas mudanças no desenvolvimento de um ser. E lá vem de novo o avanço científico, de como hoje em dia sabemos os detalhes, as causas e curas, as prevenções, e tudo o mais.
Contei também com um enriquecimento de informações. Muitos dos casos pesquisados neste trabalho eu não sabia ou sabia por alto, sem ter o conhecimento, muitas vezes, das suas principais características. E essas, me referindo às síndromes, na maioria das vezes são estéticas, o que de primeira nos causa um impacto, seguido de um olhar curioso. Pior são aquelas prejudiciais à saúde do indivíduo, o que inconscientemente nos desperta um sentimento de compaixão, consentimento instantâneo.
No fim da pesquisa, pude notar as diversidades dos casos discorridos. A riqueza de detalhes que é a biologia, a biotecnologia, a medicina, a vida, num todo. Às vezes, detalhes complexos demais para um entendimento amplo e total, mas capaz de mesmo assim nos proporcionar um acréscimo no conhecimento, o que com toda a certeza foi o objetivo principal do portfólio.
E como se não bastasse tudo isso, é notório a todos que ainda há muito o que se descobrir. A medicina tem muito a evoluir, os cientistas a se aprimorar, a genética, a ser mais utilizada e aproveitada...
Foi certamente um trabalho produtivo e os assuntos pesquisados, úteis para o nosso entendimento de vida .
Clonagem de organismos
A clonagem (do grego Klon = broto vegetal) é processo natural ou artificial onde são produzidos organismos geneticamente idênticos. Trata-se de um tipo de reprodução assexuada pois não envolve troca de gametas entre indivíduos.
Aplicações
Os cientistas têm muitas esperanças com relação à clonagem na cura de doenças, porem esbarram em parâmetros éticos. Mas acreditam que no futuro a clonagem possa produzir células de órgãos ou até órgãos inteiros, salvando a vida de muitas pessoas e diminuindo a fila dos transplantes. Que também possa utilizar células do próprio organismo no lugar de implantes mamários, clonando as células de gordura, por exemplo. A clonagem de seres humanos poderá solucionar os casos de infertilidade e até evitar que crianças nasçam com defeitos genéticos. Espécies de animais com risco de extinção podem ser clonados.
Terapia Gênica
É a introdução de um gene em tecido somático, cujo produto pode aliviar o defeito causado pela perda ou mau funcionamento de um gene vital ou de seu respectivo produto. Para o sucesso da Terapia Gênica, é necessário dois importantes fatores: (a) que não ocorra efeitos indesejáveis, e (b) que se mantenha a produção, em níveis desejáveis, do produto do gene introduzido.
A Terapia Gênica tem se expandido, podendo ser empregada em males como o câncer, AIDS e doenças neurológicas, como o mal de Parkinson e de Alzheimer, entre outros. Trataremos disto posteriormente, no item Indicações da Terapia Gênica.
Vetor de DNA
Um vetor é uma molécula de DNA em que o fragmento de DNA a ser clonado é ligado ao DNA de uma célula. Um vetor pode, por exemplo, carregar consigo um gene resistente a antibióticos, replicar-se com autonomia, e possuir uma sequência conhecida por endonuclease.
Aplicações
Os cientistas têm muitas esperanças com relação à clonagem na cura de doenças, porem esbarram em parâmetros éticos. Mas acreditam que no futuro a clonagem possa produzir células de órgãos ou até órgãos inteiros, salvando a vida de muitas pessoas e diminuindo a fila dos transplantes. Que também possa utilizar células do próprio organismo no lugar de implantes mamários, clonando as células de gordura, por exemplo. A clonagem de seres humanos poderá solucionar os casos de infertilidade e até evitar que crianças nasçam com defeitos genéticos. Espécies de animais com risco de extinção podem ser clonados.
Terapia Gênica
É a introdução de um gene em tecido somático, cujo produto pode aliviar o defeito causado pela perda ou mau funcionamento de um gene vital ou de seu respectivo produto. Para o sucesso da Terapia Gênica, é necessário dois importantes fatores: (a) que não ocorra efeitos indesejáveis, e (b) que se mantenha a produção, em níveis desejáveis, do produto do gene introduzido.
A Terapia Gênica tem se expandido, podendo ser empregada em males como o câncer, AIDS e doenças neurológicas, como o mal de Parkinson e de Alzheimer, entre outros. Trataremos disto posteriormente, no item Indicações da Terapia Gênica.
Vetor de DNA
Um vetor é uma molécula de DNA em que o fragmento de DNA a ser clonado é ligado ao DNA de uma célula. Um vetor pode, por exemplo, carregar consigo um gene resistente a antibióticos, replicar-se com autonomia, e possuir uma sequência conhecida por endonuclease.
Organismos Transgênicos
São organismos que recebem e incorporam genes de outra espécie podendo transmiti-los à sua prole. Como exemplo, temos: genes humanos em bactérias, genes de outras espécies animais e vegetais também podem ser transmitidos para espécies diferentes das espécies doadoras destes.
Quatro exemplos desses organismos são: a soja, a maça, o abacaxi e o Chester.
Há vários benefícios em torno dos alimentos transgênicos como maior resistência a pragas, maior teor proteico ou vitamínico, melhor adaptação a determinadas condições ambientais. Bons exemplos são os produtos de uso farmacêutico, como a produção de hormônios e outras proteínas humanas por bactérias ou que estão presentes no leite de animais. E outra vantagem é que esses produtos são baratos e seguros.
Porém como em tudo há riscos nas experiências com transgênicos também ocorreu na década de 80, quando fora fitos experimentos com o fator VIII de coagulação sanguínea produzido por baterias, evitando assim a sua obtenção a partir de sangue humano, porém essa prática resultou na extensa contaminação de hemofílicos pelo vírus da Aids.
Organismos Geneticamente Modificados (OGM)
Entende-se por organismo geneticamente modificado (OGM) todo o organismo cujo seu material genético foi manipulado de modo a favorecer alguma característica desejada.
Normalmente quando se fala em Organismos geneticamente modificados refere-se aos organismos transgênicos, mas estes não são exatamente a mesma coisa. Um transgénico é um organismo geneticamente modificado, mas um organismo geneticamente modificado não é obrigatoriamente um transgênico.
Um OGM é um organismos cujo material genético foi manipulado e um transgénico é um organismo que possui um ou mais genes (uma porção de DNA que codifica uma ou mais proteínas) de outro organismo no seu material genético, ou seja, uma bactéria, por exemplo, pode ser modificada geneticamente para expressar mais vezes uma proteína, mas não é um transgénico, já que não recebeu nenhum gene de outro ser vivo.
Em síntese, um organismo geneticamente modificado só é considerado um transgénico se for introduzido no seu material genético parte de material genético de outro ser.
Alguns exemplos de OGM:
- Algodão (Gossypium hirsutum)

Genes Introduzidos:
- CrylA (endotoxina) da bactéria Bacillus thuringiensis;
- Nitrilase (enzima) da bactéria Klebsiella pneumonize.
- Banana (Musa acuminata Colla)

Gene Introduzido:
- LT-B codifica (proteina enterotoxigénica) da bactéria Escherichia colli.
- Tomate (Lycopersicum esculentum)
 Gene Introduzido:
Gene Introduzido:
- Poligalacturonase (enzima) da própria planta.
Engenharia Genética
A engenharia genética consiste em modernas técnicas da biologia molecular. Tais técnicas têm a capacidade de identificar, manipular e duplicar partes dos genes dos seres vivos.
A Engenharia Genética está comprometida com algumas pesquisas, como: Indústria Farmacêutica ( vacinas; reagentes de diagnóstico; agentes anti-tumorais; novas drogas) e Agropecuária ( animais e plantas com resistência a doenças; aumento do valor nutricional de animais e plantas; plantas resistentes a pragas; produção de espécies raras ).
- Enzimas de restrição
São enzimas existentes em bactérias que tem capacidade de cortar a dupla hélice da molécula de DNA em pontos específicos, sendo por isso, chamada de “tesouras moleculares”.
Estas enzimas associam-se em uma seqüência específica de bases da molécula de DNA, quatro ou seis pares de bases, cortando a molécula nesse ponto. Esta seqüência é conhecida como palíndromo, pois as duas cadeias apresentam a mesma informação, mas em posições opostas.
Hoje, conhecemos centenas de enzimas de restrição diferentes, capazes de cortar a molécula de DNA em um determinado ponto específico.
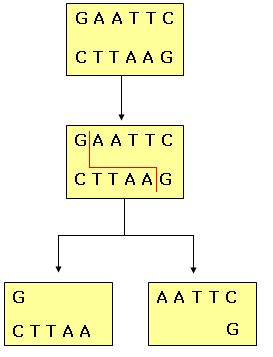
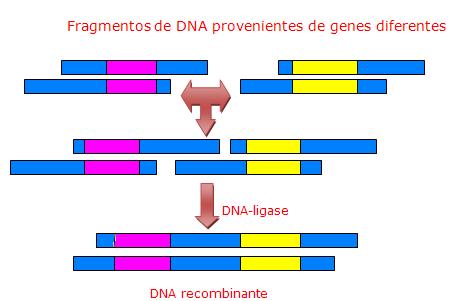
- O DNA recombinante : A Engenharia Genética criou o DNA recombinante, que nada mais é que uma seqüência de DNAs que estão combinados, mas que são de origens diferentes.
Endogamia
Endogamia é um sistema em que os acasalamentos se dão entre indivíduos aparentados, relacionados pela ascendência, ou seja, é a união de indivíduos mais aparentados do que a média da população.
Tem como efeito genético a diminuição da heterozigose e o aumento da homozigose, e, como efeito fenotípico, uma grande manifestação de genes recessivos, que acabam resultando em perda de vigor, assim como a perda da variância, à medida que aumenta o parentesco.
Endogamia pode estar estabelecida em grupos sociais, áreas ou relações. Vários tipos de endogamia foram observados em populações humanas, tal como:
- Endogamia de aldeia (por exemplo, nos Ianomâmis)
- Endogamia de linhagem (por exemplo, em comunidades pastorais do Médio Oriente)
- Endogamia de castas (por exemplo, na Índia)
- Endogamia de classes (por exemplo, nos Estados Unidos)
Melhoramento genético
O melhoramento genético é uma ciência utilizada em plantas e animais que visa aumentar a frequência de alelos favoráveis em uma população animal ou vegetal.
Para que seja iniciado um programa de melhoramento é necessário haver variabilidade genética na população, e o progresso do programa será maior tanto quanto for maior essa variabilidade.
O melhoramento animal tem por finalidade aperfeiçoar a produção dos animais que apresentam interesse para o homem. Sabe-se que o fenótipo de um indivíduo nada mais é que o produto da interação genótipo e meio ambiente, e apesar de ter a fundamentação teórica desenvolvida há muitos anos, tem recentemente, recebido grandes contribuições que são, com a necessidade de melhoria genética imposta pelo mercado, as principais responsáveis tanto pela expansão quanto pelos progressos genéticos que têm sido observados nas mais diferentes espécies de animais domésticos explorados comercialmente. Contudo, a melhoria genética se processa com base na escolha correta daqueles que participam, ou melhor, daqueles aos quais é dada a possibilidade de participar, do processo de constituição da geração seguinte. Isso vale para a escolha dos indivíduos que produzirão filhos, ou mesmo, para escolha de raças, sendo assim chamado à escolha dos indivíduos, de processo de seleção, que é importante para melhoria de raças puras ou para cruzamentos; e a escolha de raças propriamente dita, que orienta e sinaliza o sucesso dos cruzamentos. Quando se usa cruzamento, ou seja, acasalamento entre animais de raças diferentes, é importante considerar-se a heterose, também denominada de choque de sangue ou vigor híbrido. A heterose nada mais é do que a resposta obtida ao se cruzar duas ou mais raças geneticamente diferentes, tentando-se aproveitar, ao máximo, o potencial genético dos animais envolvidos, buscando-se a maior produtividade possível.

O caso de Frank Lentini
Frank Lentini (1871-1934) foi um cidadão italiano notório por ter nascido com três pernas, dois órgãos genitais e um pé no joelho da terceira perna, vindo então se tornar uma célebre atração de circos do tipo show de aberrações. Assim, no total, ele tinha três pernas, quatro pés, dezesseis dedos dos pés, e dois órgãos genitais funcionais que eram tudo o que restou de um gêmeo parasita.
Frank Lentini tinha literalmente 3 pernas. Ele nasceu em 1889 em Siracusa (Sicília) em uma família com onze filhos. Foi levado por sua tia, ainda bebê, para uma orfanato de inválidos, depois que seus pais recusaram-se a reconhecê-lo como filho.
|

Os médicos decidiram pela não remoção dos órgãos pois poderia resultar em paralisia e até a morte. Quando tinha nove anos de idade Frank deixou o orfanato de crianças inválidas na qual viveu certo período e foi levado para os EUA para ser exibido em circos de aberrações.

Apesar das adversidades, Frank nunca foi uma pessoa ressentida com sua deformidade, ele se mostrava orgulhoso por sua terceira perna e via nessa condição uma vantagem e não uma infelicidade. Estranhamente a única coisa que o incomodava em seu corpo era um dedo polegar extra que apareceu em um joelho de uma de suas pernas, Frank sempre procurou esconder esse dedão.

Em 1930 Frank Lentini se tornou oficialmente um cidadão americano, nessa mesma década conheceu Thereza Murray, paixão fulminante que terminou em casmento que gerou quatro filhos.
Foi reconhecido como um exímio jogador e conseguiu ganhar algum dinheiro com suas apresentações para os atletas das ligas de futebol. Seus filhos o descreveram como um pai presente, atencioso e muito amoroso, mas que no fundo sempre carregou certa tristeza por ter sido um dia rejeitado por seus próprios pais.

Lentini morreu na sua casa em Jacksonville, Flórida no dia 22 de setembro de 1966.

Leia mais em: Frank Lentini, o tripé humano - Metamorfose Digital http://www.mdig.com.br/index.php?itemid=7647#ixzz21gW7Vbzy
Fenilcetonúria
A fenilcetonúria (PKU) é uma doença rara na qual o bebê nasce sem a habilidade de quebrar adequadamente um aminoácido chamado fenilalanina.
Causas
A fenilcetonúria é hereditária, isto é, passa de pais para filhos. O pai e a mãe devem passar o gene defeituoso para que o bebê tenha essa doença. Isso é conhecido como traço recessivo autossômico.
Os bebês com PKU não possuem uma enzima chamada fenilalanina hidroxilase, necessária para quebrar um aminoácido essencial denominado fenilalanina. Essa substância é encontrada em alimentos que contêm proteínas.
Sem essa enzima, os níveis de fenilalanina e de duas substâncias associadas a ela crescem no organismo. Tais substâncias são prejudiciais ao sistema nervoso central e causam dano cerebral.

Sintomas
A fenilalanina atua na produção de melanina, o pigmento responsável pela cor da pele e do cabelo. Portanto, bebês com essa doença geralmente posuem pele, cabelo e olhos mais claros do que seus irmãos que não sofrem dessa doença.
Se a doença não for tratada ou se os alimentos contendo fenilalanina não forem evitados, um odor "de rato" poderá ser sentido no hálito, na pele e na urina. Esse odor incomum deve-se ao aumento de substâncias de fenilalanina no organismo.
Tratamento
A PKU é uma doença tratável. O tratamento consiste em uma dieta extremamente baixa em fenilalanina, especialmente durante o crescimento da criança. A dieta deve ser rigorosamente seguida. Isso demanda uma supervisão apurada de um nutricionista ou de um médico, além da cooperação dos pais e da criança. Aqueles que mantêm a dieta durante a vida adulta possuem saúde física e mental melhores. "Dieta para sempre" tornou-se uma bandeira levantada pela maioria dos especialistas. Isso é muito importante, principalmente antes da concepção e durante a gestação.
A fenilalanina é encontrada significantemente em leite, ovos e outros alimentos comuns. Adoçantes com aspartame também contém fenilalanina. Todos os produtos que contém aspartame devem ser evitados.
Uma fórmula especial chamada Lofenalac é feita para crianças com PKU. Ela pode ser usada por toda a vida como uma fonte de proteína extremamente pobre em fenilalanina e equilibrada para os aminoácidos essenciais restantes.
O uso de suplementos como óleo de peixe para substituir a longa cadeia de ácidos gordurosos ausentes em uma dieta padrão sem fenilalanina pode ajudar a melhorar o desenvolvimento neurológico, inclusive a coordenação motora fina. Outros suplementos específicos, como ferro ou carnitina, podem ser necessários.
Galactosemia
A galactosemia é um distúrbio no qual o corpo não consegue transformar (metabolizar) galactose em glicose.
A galactosemia é uma doença hereditária. É passada de geração em geração.
Ocorre, aproximadamente, em 1 a cada 60.000 partos de indivíduos de pele branca. A taxa é diferente em outros grupos.
Existem três formas da doença: a deficiência da enzima galactose1fosfato uridil transferase (galactosemia clássica, a forma mais comum e mais grave), a deficiência de galactoquinas e a deficiência de galactose-6-fosfato epimerase.
As pessoas com galactosemia não conseguem transformar o açúcar simples da galactose. A galactose compõe metade da lactose, o açúcar encontrado no leite. O outro açúcar é a glicose.
Se um bebê com galactosemia tomar leite, substâncias feitas de galactose se acumularão em seu sistema. Essas substâncias danificam o fígado, cérebro, rins e olhos.
Pessoas com galactosemia não toleram qualquer tipo de leite (humano ou animal). Elas devem ter cuidado com a ingestão de outros alimentos que contenham galactose.
SINTOMAS DE GALACTOSEMIA
Bebês com galactosemia podem desenvolver sintomas nos primeiros dias de vida se consumirem leite materno ou qualquer outro alimento que contenha lactose. Os sintomas podem ser devido a uma infecção grave no sangue com a bactéria E. coli.
- Convulsões
- Irritabilidade
- Letargia
- Má alimentação (o bebê se recusa a tomar mamadeira com leite em pó)
- Ganho de peso insuficiente
- Pele e olhos amarelados (icterícia)
- Vômitos
TRATAMENTO
As pessoas com esse problema devem evitar todo tipo de leite e produtos que contenham leite (inclusive leite em pó), e outros alimentos que contenham galactose por toda a vida. É essencial ler os rótulos dos produtos e ser um consumidor bem informado.
Hipotiroidismo congênito
De acordo com Brasil (2002), Hipotireoidismo Congênito ocorre quando a glândula tireóide do recém-nascido (RN) não é capaz de produzir quantidades adequadas de hormônios tireoidianos, o que resulta numa redução generalizada dos processos metabólicos.
A patologia pode ser classificada em:
• Primária – quando a falha ocorre na glândula tireóide;
• Secundária – quando ocorre deficiência do TSH hipofisário;
• Terciária – quando ocorre deficiência do TRH hipotalâmico;
• Resistência periférica à ação dos hormônios tireóideos.
Em regiões onde a deficiência de iodo não é endêmica, o Hipotiroidismo Congênito é mais freqüentemente causado pela glândula tireóide ausente ou ectópica (Hipotiroidismo Primário), de etiologia esporádica. Mais raramente, em cerca de 15% dos casos, é uma patologia herdada recessivamente, levando a uma falha na biossíntese do hormônio tireoidiano.
Em crianças não submetidas a programas de Triagem Neonatal e, conseqüentemente, não tratadas precocemente, o crescimento e o desenvolvimento mental ficam seriamente comprometidos. As manifestações clínicas são: hipotonia muscular, dificuldades respiratórias, cianose, icterícia prolongada, constipação, bradicardia, anemia, sonolência excessiva, livedo reticularis, choro rouco, hérnia umbilical, alargamento de fontanelas, mixedema, sopro cardíaco, dificuldade na alimentação com deficiente crescimento pôndero-estatural, atraso na dentição, retardo na maturação óssea, pele seca e sem elasticidade, atraso de desenvolvimento neuropsicomotor e retardo mental.
As crianças que realizam diagnóstico precoce através dos programas de Triagem Neonatal não apresentam qualquer sintomatologia clínica, desde que a terapia de reposição hormonal seja iniciada precocemente.
O momento ideal para o diagnóstico do Hipotireoidismo Congênito é o período neonatal, pois se sabe que a partir de 4 semanas de vida, a deficiência de hormônios tireóideos já pode causar alguma lesão neurológica.
A triagem pode perder raros casos de Hipotireoidismo Congênito, tais como Hipotireoidismo Pituitário Hipotalâmico, doença compensada (T4 normal, TSH elevado) ou aumento de TSH tardio, que são muito raros ( talvez 2 a 3 por 100.000).
Sempre deve ser realizada a dosagem de T4 (T4 total e T4 livre) e TSH em amostra de sangue venoso, obtida o mais cedo possível após os resultados positivos iniciais no Programa de Triagem Neonatal, para que haja a confirmação diagnóstica. Agindo dessa forma, a média de detecção dos casos suspeitos é de aproximadamente 90%. Os 10% dos casos restantes são menos severamente afetados e não se tornam detectáveis por TSH até a idade de 2 a 6 semanas.
Para que seja determinada a etiologia do processo, como na maioria das vezes (85%), a origem é na própria glândula tireóide, existe a indicação de realizar exames de ultrassonografia da tireóide ou cintilografia com captação tireóidea de iodo radioativo. Quando a espera para a feitura desses exames puder vir a retardar o início da terapia de reposição hormonal, os mesmos serão deixados para serem realizados somente após os 2 anos de vida da criança, quando poderemos suspender a medicação para sua realização. Nos casos mais raros de etiologia secundária ou terciária, indicam-se também os testes laboratoriais com estímulo de TRH.
Tratamento
O tratamento da patologia consiste na reposição dos hormônios tireóideos deficitários, no caso, reposição de Levotiroxina. A Levotiroxina Sódica é o sal sódico do isômero sintético da Tiroxina (T4), sendo que sua utilização para reposição hormonal produz a normalização do estado metabólico que se encontra deficiente no Hipotiroidismo. No meio intracelular, T4 é convertido em T3, dessa forma disponibiliza-se ambos os hormônios tireóideos, mesmo administrando somente um deles. O tratamento preconizado deverá ser mantido por toda a vida.
A Levotiroxina é apresentada na forma de comprimidos que contém 25 a 300 µg, e na forma de pó para reconstituição para uso em injeções, sendo que a dose utilizada varia de acordo com a idade do paciente e seu peso corporal, sendo que as crianças mais jovens necessitam doses superiores às crianças maiores e aos adultos. Inicia-se calculando doses de 10 a 15 µg/Kg/dia, para o RN a termo, após isso, a dose é recalculada conforme o ganho ponderal da criança e os níveis de T4 e TSH observados nos controles laboratoriais subseqüentes.
A meia-vida da Levotiroxina é de sete dias, sendo então administrada somente uma vez ao dia. Apresenta boa absorção via oral, havendo raramente a necessidade de sua utilização por via parenteral (neste caso, utiliza-se 75 a 80% da dose preconizada via oral).
Teste do pezinho
Os bebês são submetidos a uma bateria de exames logo quando nascem, com o intuito de identificar quaisquer anormalidades e prevenir uma série de doenças. A triagem neonatal, mais conhecida como teste do pezinho, é um dos exames mais importantes na hora de detectar irregularidades na saúde da criança.
Com apenas algumas gotas de sangue colhidas do calcanhar do recém-nascido, o teste permite diagnosticar precocemente oito doenças, entre metabólicas, congênitas e infecciosas. A triagem deve ser feita entre o terceiro e o sétimo dia de vida do bebê, já que antes disso os resultados podem não ser muito precisos.
O teste do pezinho chegou ao Brasil na década de 70 para identificar a fenilcetonúria e o hipotireoidismo congênito. Em 1992, o teste se tornou obrigatório em todo o território nacional.

Conheça as oito doenças identificadas no teste do pezinho:
Anemia falciforme – doença hereditária que altera a formação da hemoglobina, molécula responsável pelo transporte do oxigênio no sangue. Em decorrência dessa alteração, as hemácias ficam com forma de foice (daí o nome “falciforme”), o que dificulta sua locomoção e acaba lesionando tecidos.
Deficiência de biotionidase – é a falta da vitamina biotina no organismo. Sua deficiência resulta em convulsões, fraqueza muscular, queda de cabelo, surgimento de espinhas, acidez do sangue e baixa imunidade.
Fenilcetonúria – é uma doença genética caracterizada pela incapacidade de metabolizar a enzima fenilalanina, responsável pela produção do aminoácido tirosina. A ausência de tirosina pode acarretar retardação mental.
Galactosemia – é uma doença genética que dificulta a conversão de galactose (açúcar presente no leite) em glicose. O resultado é o acúmulo de galactose no organismo, causando problemas de coagulação, icterícia (pele amarelada), hipoglicemia (baixa da taxa de glicose no sangue), glicosúria (excesso de glicose na urina), acidez do sangue e catarata.
Glicose 6-fosfato desidrogenase – distúrbio metabólico que causa alterações das enzimas fundamentais para proteção das células, especialmente das hemácias. Sem estabilidade, os glóbulos vermelhos podem morrer, causando anemia hemolítica.
Hipotireoidismo congênito – doença que faz com que a glândula tireoide não seja capaz de produzir quantidade adequada de hormônios tireoidianos, o que deixa os processos metabólicos mais lentos. Uma das principais consequências é a retardação mental.
Hiperplasia congênita da supra-renal – provoca uma deficiência na produção de hormônios pelas glândulas supra-renais ou adrenais. Para compensar, a hipófise produz excesso de hormônios que estimulam as supra-renais, que aumentam de tamanho e passam a produzir em excesso hormônios que levam à masculinização do corpo da criança. Além disso, pode ocorrer desidratação, perda de sal no organismo e vômitos.
Toxoplasmose – é uma doença infecciosa causada pelo parasita Toxoplasma gondii, que pode causar calcificações cerebrais, malformações, doença sistêmica grave. Tardiamente, pode se expressar causando doenças da retina.
Talassemia
Talassemia, também conhecida como anemia do Mediterrâneo, é uma doença hereditária trazida para o Brasil pelos habitantes dos países banhados pelo mar Mediterrâneo (portugueses, espanhóis, italianos, gregos, egípcios, libaneses). Sua principal característica é a produção anômala de hemoglobina, uma proteína do sangue responsável pelo transporte de oxigênio para todos os tecidos do organismo.
Existem dois tipos de talassemia – alpha e beta – que podem manifestar-se nas seguintes formas: minor, intermediária e major. A forma minor, ou traço talassêmico, produz um grau de anemia leve, assintomático e que pode passar totalmente despercebido. Na forma intermediária, a deficiência da síntese de hemoglobina é moderada e as consequências menos graves. Já a talassemia major, ou anemia de Cooley, é uma forma grave da doença, causada pela transmissão de dois genes defeituosos, um do pai e outro da mãe. Isso provoca anemia profunda e outras alterações orgânicas importantes, como o aumento do baço, atraso no crescimento e problemas nos ossos.
Sintomas
Os sintomas estão diretamente relacionados com a gravidade da doença. Os mais comuns são: cansaço e fraqueza; palidez e icterícia; atraso no crescimento; abdômen desenvolvido; aumento do baço; e alterações ósseas.
Diagnóstico
Para estabelecer o diagnóstico, é importante levantar a história clinica e obter informações sobre a origem étnica do paciente. Exames de laboratório, entre eles a eletroforese de hemoglobina quantitativa e qualitativa, são importantes para determinar o tipo da doença.
Tratamento
A talassemia minor não demanda tratamento específico. Em certas circunstâncias (na gravidez, por exemplo), a suplementação com acido fólico pode trazer benefícios para os portadores da doença.
A talassemia intermediária pode requerer a indicação de transfusões de sangue com a finalidade de aumentar a oferta de glóbulos vermelhos.
Portador de talassemia major necessita de transfusões de sangue regulares e de medicamentos para retirar o excesso de ferro que se acumula em determinados órgãos (terapia quelante do ferro). O transplante de medula óssea também pode constituir uma solução terapêutica nesses casos.
Hemofilia
Hemofilia é uma doença genético-hereditária que se caracteriza por desordem no mecanismo de coagulação do sangue e manifesta-se quase exclusivamente no sexo masculino.
Existem dois tipos de hemofilia: A e B. A hemofilia A ocorre por deficiência do fator VIII de coagulação do sangue e a hemofilia B, por deficiência do fator IX.
A doença pode ser classificada, ainda, segundo a quantidade do fator deficitário em três categorias: grave (fator menor do que 1%), moderada (de 1% a 5%) e leve, acima de 5%. Neste caso, às vezes, a enfermidade passa despercebida até a idade adulta.
O gene que causa a hemofilia é transmitido pelo par de cromossomos sexuais XX. Em geral, as mulheres não desenvolvem a doença, mas são portadoras do defeito. O filho do sexo masculino é que pode manifestar a enfermidade.

Diagnóstico
Além dos sinais clínicos, o diagnóstico é feito por meio de um exame de sangue que mede a dosagem do nível dos fatores VIII e IX de coagulação sanguínea.
Sintomas
Nos quadros graves e moderados, os sangramentos repetem-se espontaneamente. Em geral, são hemorragias intramusculares e intra-articulares que desgastam primeiro as cartilagens e depois provocam lesões ósseas. Os principais sintomas são dor forte, aumento da temperatura e restrição de movimento. As articulações mais comprometidas costumam ser joelho, tornozelo e cotovelo.
Os episódios de sangramento podem ocorrer logo no primeiro ano de vida do paciente sob a forma de equimoses (manchas roxas), que se tornam mais evidentes quando a criança começa a andar e a cair. No entanto, quando acometem a musculatura das costas, não costumam exteriorizar-se.
Nos quadros leves, o sangramento ocorre em situações como cirurgias, extração de dentes e traumas.
Tratamento
O tratamento da hemofilia evoluiu muito e, basicamente, consiste na reposição do fator anti-hemofílico. Paciente com hemofilia A recebe a molécula do fator VIII, e com hemofilia B, a molécula do fator IX. Os hemocentros distribuem gratuitamente essa medicação que é fornecida pelo Ministério da Saúde.
Quanto mais precoce for o início do tratamento, menores serão as seqüelas que deixarão os sangramentos. Por isso, o paciente deve ter em casa a dose de urgência do fator anti-hemofílico específico para seu caso e ser treinado para aplicá-la em si mesmo tão logo apareçam os primeiros sintomas.
Deve também fazer também aplicações de gelo, no mínimo, três vezes por dia, por 15 ou 20 minutos, até que a hemorragia estanque.
Vencida a fase aguda, o portador de hemofilia deve ser encaminhado para fisioterapia a fim de reforçar a musculatura e promover estabilidade articular.
Doença de Alzheimer


O diagnóstico de Doença de Alzheimer é feito através da exclusão de outras doenças que podem evoluir também com quadros demenciais. Por exemplo: tumores cerebrais, depressão, hidrocefalia, etc.
Doença de Parkinson
Doença neurológica, sem causa conhecida, que atinge o sistema nervoso central e compromete os movimentos. Quanto maior a faixa etária, maior a incidência da doença de Parkinson. De acordo com as estatísticas, na grande maioria dos pacientes, ela surge a partir dos 55, 60 anos e sua prevalência aumenta a partir dos 70, 75 anos.
Sintomas
Os sintomas da doença de Parkinson variam de um paciente para o outro. Em geral, no início, eles se apresentam de maneira lenta, insidiosa, e o paciente tem dificuldade de precisar a época em que apareceram pela primeira vez.
A lentificação dos movimentos e os tremores nas extremidades das mãos, muias vezes notados apenas pelos amigos e familiares, costumam ser os primeiros sinais da doença. A diminuição do tamanho das letras ao escrever é outra característica importante.
Outros sintomas podem estar associados ao início da doença: rigidez muscular; acinesia (redução da quantidade de movimentos), distúrbios da fala, dificuldade para engolir, depressão, dores, tontura e distúrbios do sono, respiratórios, urinários.
Tratamento
O tratamento pode ser medicamentoso, psicoterápico e até cirúrgico em alguns casos.
O tratamento medicamentoso é feito à base de drogas neuroprotetoras que visam a evitar a diminuição progressiva de dopamina, neurotransmissor responsável pela transmissão de sinais na cadeia de circuitos nervosos.
O tratamento psicoterápico ocorre em função da depressão, perda de memória e do aparecimento de demências e pode incluir a prescrição de medicamentos antidepressivos e de outros psicotrópicos.

Doença de Tay-Sachs
É uma enfermidade causada pela disfunção dos lisossomos, organelas responsáveis pela digestão celular. Resulta de um defeito na hexosaminidase A, enzima que catalisa uma das etapas da digestão intracelular de um lipídio abundante nas membranas das células nervosas, o gangliosídio.
Trata-se de uma doença oriunda de uma herança autossômica recessiva, ou seja, o indivíduo só desenvolve a doença quando herdar os genes defeituosos tanto do pai quanto da mãe. Aqueles que recebem os genes recessivos de apenas um dos genitores não desenvolvem a doença, mas são portadores de genes mutados, e caso tenha filhos com outro portador, os filhos terão a doença de Tay-sachs.
Os sintomas da doença começam a se manifestar ainda no primeiro ano de vida do indivíduo. Por ser uma doença neurodegenerativa, a criança tem seu sistema nervoso bastante comprometido, principalmente no que concerne à capacidade psicomotora. Um dos sinais mais característicos da doença de Tay-Sachs é o aparecimento de uma mancha vermelha no olho, seguida de cegueira, surdez, incapacidade de engolir, atrofia dos músculos e paralisia. Em alguns casos, essa mancha pode não aparecer no início do quadro, o que não descarta o diagnóstico da doença, visto que ela pode surgir posteriormente.

Por volta dos dois anos de idade, a criança já apresenta sinais de demência, que consiste numa perda das capacidades cognitivas, tais como percepção, atenção, linguagem, memória, raciocínio, pensamento, imaginação. As células nervosas dos portadores da doença incham devido ao acúmulo de gangliosídios não digeridos, provocando um aumento expressivo do crânio. A doença evolui irreversivelmente ao óbito do indivíduo, que se dá, geralmente, até os 5 anos de vida.
O diagnóstico da doença de Tay-Sachs é dado por meio de um teste laboratorial, em que são determinados os níveis de hexosaminadase A no sangue. Os portadores da enfermidade apresentam uma significativa redução da quantidade e da atividade da enzima. Os sinais clínicos e o histórico familiar muito contribuem para o diagnóstico.
A doença também pode ser detectada durante a gestação quando há a certeza de que os pais são portadores. Para isso, é feita a biópsia das vilosidades coriônicas (estruturas presentes na placenta), exame que possibilita a identificação de genes mutantes do feto. Muitos casais, aos descobrirem que o feto é portador da doença, optam por interromper a gestação.
Não existe cura para a doença de Tay-Sachs, e sim tratamentos e medidas paliativas para os sintomas. Os portadores da doença, em geral, fazem uso de medicamentos anticonvulsionantes, que diminuem as crises de convulsões. O transplante de medula óssea pode ser indicado, pois a introdução de células sadias no organismo acarretará na produção de enzimas totalmente ativas. É possível, também que o indivíduo passe por uma terapia gênica, ou geneterapia, em que novos genes serão inseridos nas células somáticas, que passarão a produzir a enzima em quantidades suficientes. Embora promissora, a geneterapia é ainda é um tratamento em pesquisa.
Cariótipo

Itabaianinha (Terra dos anões)
Isso mesmo, o clássico da Wall Disney possui uma versão brasileira em Sergipe, mais realista e poética. A 120 km de Aracaju, Itabaianinha, com 32.00 habitantes, possui cerca de 80 itabaianinhenses adultos com menos de 1,30 metro. Há pelo menos 200 anos, é comum ver uma porrada de anões caminhando pela cidade. E detalhe, o motivo é por causa do incesto inevitável.

Itabaianinha causou tanto interesse que foi tema do documentário Terra de Gigantes , de Ana Paula Teixeira, e hoje é famosa por receber cientistas norte americanos em busca de explicações para o caso. Existe inclusive uma ministração de hormônios aos anões mais novos para que voltem a crescer. Porém, há aqueles que recusam o medicamento e preferem continuar carregando apelidos: Zé Miúdo, Mundinho, Joaninha, Lerinho. Essa é a Itabaianinha, a cidade do "inho".
Síndrome de Treacher Collins
É um distúrbio do desenvolvimento craniofacial de herança autossômica dominante. Cerca de 60% das pessoas afetadas são decorrentes de mutações novas, isto é, seus pais não são afetados. Uma pessoa afetada têm 50% de probabilidade de transmitir a mutação e assim ter uma criança também afetada.
A síndrome de Treacher Collins é causada por mutações no gene TCOF1 (cromossomo 5), que tem 26 éxons e codifica uma proteína chamada treacle. Esta tem funções importantes na manutenção das células derivadas da crista neural (células que vão formar os ossos do ouvido, face e também as orelhas) durante as primeiras semanas de desenvolvimento do embrião. As mutações patogênicas em geral são específicas para cada paciente e estão localizadas ao longo do gene, ou seja não há ponto quente de ocorrência destas mutações.
O teste genético para a detecção de mutações no gene TCOF1 pode confirmar o diagnóstico clínico e é particularmente importante em casos com quadro clínico leve, casos em que há dúvidas no diagnóstico e quando há apenas um indivíduo afetado na família.
Características
A síndrome de Treacher Collins é caracterizada por achatamento dos ossos malares da face (hipoplasia malar), queixo pequeno (micrognatia), orelhas pequenas, mal-formadas ou ausentes, surdez total ou parcial, defeitos nas pálpebras inferiores (coloboma), olhos com os cantos externos “caídos” para baixo e palato estreito ou fissurado.
Existe uma grande variabilidade nos sinais clínicos, mesmo entre pessoas da mesma família. Existem casos tão leves que podem passar despercebidos ao diagnóstico, e até casos graves, com surdez profunda e que necessitam de diversas cirurgias corretivas.


Síndrome do Triplo-X
É uma aberração cromossômica numérica que atinge 1 em cerca de 800 a 1000 mulheres. As mulheres portadoras dessa síndrome apresentam um cromossomo X a mais, totalizando um cariótipo com 47 cromossomos: 47, XXX. Quase todos os erros relacionados à essa síndrome ocorrem durante a ovulogênese, pela não disjunção dos cromossomos.


Algumas mulheres podem apresentar até 4 ou 5 cromossomos X extras. Quanto mais cromossomos X, maio o índice de retardo mental nessas mulheres.
Existem mulheres portadoras dessas aberrações cromossômicas, mas desconhecem a doença por não apresentarem os sintomas.
Os sintomas dessa síndrome envolvem menor grau de inteligência, múltiplas peles frouxas no pescoço, podem entrar na menopausa precocemente, retardamento mental (o grau de retardamento varia entre as portadoras), entre outras.
A doença pode ser diagnosticada por estudos do cariótipo, onde é claramente visível a presença de três cromossomos sexuais X.
Cariótipo
Síndrome do "x" frágil
As pessoas são formadas por células, cada célula possui dentro do seu núcleo o material genético que se agrupa em estruturas chamadas de cromossomos. Nos cromossos estão os genes.
Dentro de uma célula há 46 cromossomos, metade vinda do pai, metade vinda da mãe, um destes pares é chamado de cromossos sexuais: sendo XX para o sexo feminino (um X do pai e outro da mãe) e XY para o sexo masculino (o X proveniente da mãe e o Y do pai).
Assim, como trata-se da Síndrome do X Frágil, a alteração genética ocorre apenas no cromossomo X, portanto, em mulheres o quadro clínco geralmente é menos grave porque o outro X compensa cromossomo afetado. Já em homens como o outro cromossomo do par é um Y os quadros são mais severos e os pais portadores do gene alterado passarão a síndrome para todas as suas filhas, mas não o farão para nenhum de seus filhos.
Esta é uma doença genética, causada por uma expansão de trinucleotídeos CGG no primeiro exon do gene FMR-1, localizado na região Xq27.3 no cromossomo X. Entretanto esta alteração genética não ocorre em uma geração, mas acontece em etapas ao longo de gerações.
O gene FMR1 normalmente contém entre 6 e 50 repetições de CGG. Entre 50 e 200 repetições caracteriza-se estado de pré-mutação, não havendo metilação anormal e sendo o paciente portador assintomático. Um número maior do que 200 repetições confere ao paciente o estado de portador sintomático em 100% dos homens e em 50 a 70% das mulheres12,13. As pré-mutações são transmitidas pelos homens para suas filhas sem que haja novas mutações, enquanto que, quando transmitidas por mulheres, têm grandes chances de se converterem a mutações completas.
Primeiramente ocorrem pré-mutações, isto é, alterações que já fogem ao normal, mas ainda não são severas o suficiente para provocarem "sinais e sintomas" da síndrome, portanto, seus portadores não sabem que são portadores destas pré-mutações.
HISTÓRIA
A ligação entre deficiência mental e o gene sexual foi descrita primeiramente por J. Purdon Martin e Julia Bell, em 1943, no artigo "A PEDIGREE OF MENTAL DEFECT SHOWING SEX-LINKAGE", em que analisaram uma família (6 gerações), mostrando que a deficiência mental, passava de mães saudáveis (porém portadoras) para filhos, mas não para filhas.
Já havia observações clínicas quanto ao maior número de meninos com retardo mental que meninas, inclusive institucionalizados, no final do século XIX e início do século XX. Entretanto, coube a Leherke (1969) estabelecer esta relação com genes ligados ao cromossomo X. No mesmo ano Lubs descreveu uma outra família afetada e curiosamente em 1973 um trabalho brasileiro de Escalante e Frota-Pessoa que descrevia outra família não teve o "impacto merecido", possivelmente por ter sido publicado em português e no Brasil.
Acredita-se que 1 em 250 mulheres e 1:700 homens sejam portadores da pré-mutação.
A quantidade de homens afetados pela síndrome é maior que a de mulheres, mas as estimativas são muito variáveis, indo desde 1 caso para cada 1250 homens até 1:4000 e de 1:4000 a 1:6000 mulheres.
TRATAMENTO
O tratamento da síndrome baseia-se em estimulação da aquisição de aptidões (habilidades motoras, intelectuais e sociais) e no tratamento específico ou sintomático de doenças ou sintomas que possam surgir.
Assinar:
Postagens (Atom)